Nucleotide diversity#
Using REF as ancestral alleles#
We are wondering how the ancestral allele inference effect the final treesequence object.
We are testing a tstree object created by imposing the reference allele as the
ancestral allele. The effect is that there are more or less the same mutations as the
number of variants (while in the case of the ancestrall allele calculated with
est-sfs we can see million of mutations). Let’s start by loading data for this
REF-based treeseq object
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
import tskit
from tskitetude import get_project_dir
from tskitetude.helper import create_windows
ts_reference = tskit.load(str(get_project_dir() / "results-reference/background_samples/tsinfer/SMARTER-OA-OAR3-forward-0.4.10.focal.26.trees"))
ts_reference
|
|
|
|---|---|
| Trees | 659 |
| Sequence Length | 44 077 779 |
| Time Units | generations |
| Sample Nodes | 11 478 |
| Total Size | 87.9 MiB |
| Metadata |
dict |
| Table | Rows | Size | Has Metadata |
|---|---|---|---|
| Edges | 2 266 939 | 69.2 MiB | |
| Individuals | 5 739 | 353.1 KiB | ✅ |
| Migrations | 0 | 8 Bytes | |
| Mutations | 657 | 23.8 KiB | |
| Nodes | 21 091 | 1.1 MiB | ✅ |
| Populations | 202 | 4.7 KiB | ✅ |
| Provenances | 4 | 2.4 KiB | |
| Sites | 657 | 32.7 KiB | ✅ |
| Provenance Timestamp | Software Name | Version | Command | Full record |
|---|---|---|---|---|
| 06 September, 2024 at 01:19:45 AM | tsdate | 0.1.7 | inside_outside |
Detailsdictschema_version: 1.0.0
software:
dictname: tsdateversion: 0.1.7
parameters:
dictmutation_rate: 1e-08recombination_rate: None time_units: None progress: None population_size: 10000.0 eps: 1e-06 outside_standardize: True ignore_oldest_root: False probability_space: logarithmic num_threads: None cache_inside: False command: inside_outside
environment:
dict
os:
dictsystem: Linuxnode: node1 release: 5.15.0-58-generic version: #64-Ubuntu SMP Thu Jan 5 11:43:13 UTC 2023 machine: x86_64
python:
dictimplementation: CPythonversion: 3.11.9
libraries:
dict
tskit:
dictversion: 0.5.8 |
| 06 September, 2024 at 01:13:11 AM | tsdate | 0.1.7 | preprocess_ts |
Detailsdictschema_version: 1.0.0
software:
dictname: tsdateversion: 0.1.7
parameters:
dictminimum_gap: 1000000remove_telomeres: True filter_populations: False filter_individuals: False filter_sites: False
delete_intervals:
listlist0209048.0 list44004282.044077779.0 command: preprocess_ts
environment:
dict
os:
dictsystem: Linuxnode: node1 release: 5.15.0-58-generic version: #64-Ubuntu SMP Thu Jan 5 11:43:13 UTC 2023 machine: x86_64
python:
dictimplementation: CPythonversion: 3.11.9
libraries:
dict
tskit:
dictversion: 0.5.8 |
| 06 September, 2024 at 01:13:05 AM | tskit | 0.5.8 | simplify |
Detailsdictschema_version: 1.0.0
software:
dictname: tskitversion: 0.5.8
parameters:
dictcommand: simplifyTODO: add simplify parameters
environment:
dict
os:
dictsystem: Linuxnode: node1 release: 5.15.0-58-generic version: #64-Ubuntu SMP Thu Jan 5 11:43:13 UTC 2023 machine: x86_64
python:
dictimplementation: CPythonversion: 3.11.9
libraries:
dict
kastore:
dictversion: 2.1.1 |
| 06 September, 2024 at 01:13:01 AM | tsinfer | 0.3.3 | infer |
Detailsdictschema_version: 1.0.0
software:
dictname: tsinferversion: 0.3.3
parameters:
dictmismatch_ratio: Nonecommand: infer
environment:
dict
libraries:
dict
zarr:
dictversion: 2.17.2
numcodecs:
dictversion: 0.12.1
lmdb:
dictversion: 1.4.1
tskit:
dictversion: 0.5.8
os:
dictsystem: Linuxnode: node1 release: 5.15.0-58-generic version: #64-Ubuntu SMP Thu Jan 5 11:43:13 UTC 2023 machine: x86_64
python:
dictimplementation: CPython
version:
list311 9 |
I need to calculate nucleotide diversity per site. The only way to do this seems
to be calculating windows containing the SNPs an then calculating the nucleotide
diversity with the tskit.TreeSequence.diversity function. I’ve created a function
create_windows in helper module:
# the last index is a simply a 2 step starting from position 1
reference_diversity = ts_reference.diversity(windows=create_windows(ts_reference))[1::2]
reference_diversity[:10]
array([0.42881531, 0.48983504, 0.49397942, 0.27006834, 0.45935871,
0.24743806, 0.14849395, 0.3905915 , 0.46073859, 0.4897602 ])
Now let’s compare the nucleotide diversity calculated using vcftools: here’s the command line to calculate nucleotide diversity per site:
cd results-reference/background_samples/focal
vcftools --gzvcf SMARTER-OA-OAR3-forward-0.4.10.focal.26.vcf.gz --out allsamples_pi --site-pi
The allsamples_pi.sites.pi is a TSV file with the positions and the nucleotide diversity. Read it with pandas:
vcftools_diversity = pd.read_csv(get_project_dir() / "results-reference/background_samples/focal/allsamples_pi.sites.pi", sep="\t")
vcftools_diversity.head()
| CHROM | POS | PI | |
|---|---|---|---|
| 0 | 26 | 209049 | 0.428815 |
| 1 | 26 | 268822 | 0.489835 |
| 2 | 26 | 285471 | 0.493979 |
| 3 | 26 | 405330 | 0.270068 |
| 4 | 26 | 528308 | 0.459359 |
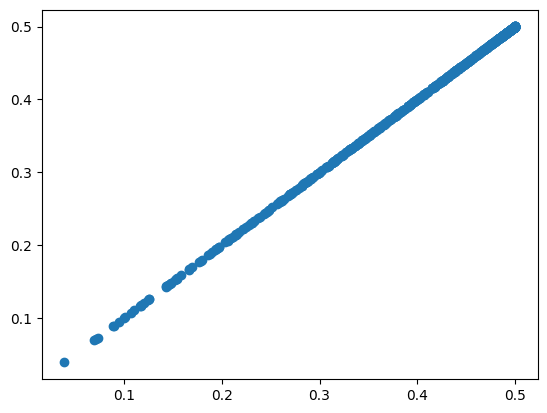
Are this values similar?
np.isclose(reference_diversity, vcftools_diversity["PI"], atol=1e-6).all()
np.True_
Calculate diversity using branch:
# the last index is a simply a 2 step starting from position 1
reference_diversity_branch = ts_reference.diversity(mode='branch', windows=create_windows(ts_reference))[1::2]
reference_diversity_branch[:10]
array([24.21275953, 24.51004717, 20.56900189, 20.18076343, 23.28547977,
17.66467911, 37.04506148, 16.55024199, 63.8544266 , 15.75780889])
Try to plot the tow different diversities with vcftools output:
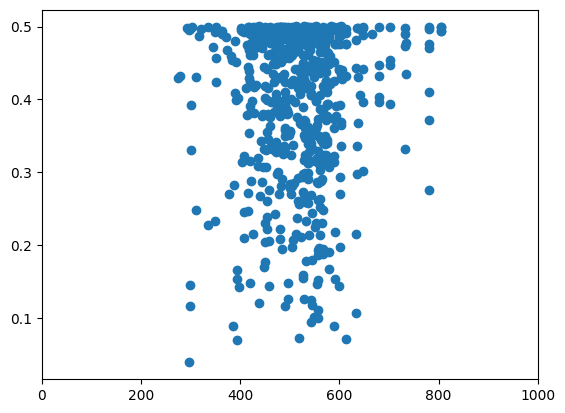
plt.scatter(reference_diversity, vcftools_diversity["PI"])
<matplotlib.collections.PathCollection at 0x7f6f3f007230>
plt.scatter(reference_diversity_branch, vcftools_diversity["PI"])
plt.xlim(0, 1000)
(0.0, 1000.0)
EST-SFS output as ancestral alleles#
Can we calculate nucleotide diversity using the tree files generated by the pipeline
using the est-sfs output as ancestral alleles?
ts_estsfs = tskit.load(str(get_project_dir() / "results-estsfs/background_samples/tsinfer/SMARTER-OA-OAR3-forward-0.4.10.focal.26.trees"))
ts_estsfs
|
|
|
|---|---|
| Trees | 539 |
| Sequence Length | 44 077 779 |
| Time Units | generations |
| Sample Nodes | 11 478 |
| Total Size | 87.5 MiB |
| Metadata |
dict |
| Table | Rows | Size | Has Metadata |
|---|---|---|---|
| Edges | 1 962 608 | 59.9 MiB | |
| Individuals | 5 739 | 353.1 KiB | ✅ |
| Migrations | 0 | 8 Bytes | |
| Mutations | 321 178 | 11.3 MiB | |
| Nodes | 19 785 | 977.3 KiB | ✅ |
| Populations | 202 | 4.7 KiB | ✅ |
| Provenances | 4 | 2.4 KiB | |
| Sites | 657 | 33.3 KiB | ✅ |
| Provenance Timestamp | Software Name | Version | Command | Full record |
|---|---|---|---|---|
| 05 September, 2024 at 11:56:43 PM | tsdate | 0.1.7 | inside_outside |
Detailsdictschema_version: 1.0.0
software:
dictname: tsdateversion: 0.1.7
parameters:
dictmutation_rate: 1e-08recombination_rate: None time_units: None progress: None population_size: 10000.0 eps: 1e-06 outside_standardize: True ignore_oldest_root: False probability_space: logarithmic num_threads: None cache_inside: False command: inside_outside
environment:
dict
os:
dictsystem: Linuxnode: node2 release: 5.15.0-58-generic version: #64-Ubuntu SMP Thu Jan 5 11:43:13 UTC 2023 machine: x86_64
python:
dictimplementation: CPythonversion: 3.11.9
libraries:
dict
tskit:
dictversion: 0.5.8 |
| 05 September, 2024 at 11:49:51 PM | tsdate | 0.1.7 | preprocess_ts |
Detailsdictschema_version: 1.0.0
software:
dictname: tsdateversion: 0.1.7
parameters:
dictminimum_gap: 1000000remove_telomeres: True filter_populations: False filter_individuals: False filter_sites: False
delete_intervals:
listlist0209048.0 list44004282.044077779.0 command: preprocess_ts
environment:
dict
os:
dictsystem: Linuxnode: node2 release: 5.15.0-58-generic version: #64-Ubuntu SMP Thu Jan 5 11:43:13 UTC 2023 machine: x86_64
python:
dictimplementation: CPythonversion: 3.11.9
libraries:
dict
tskit:
dictversion: 0.5.8 |
| 05 September, 2024 at 11:49:46 PM | tskit | 0.5.8 | simplify |
Detailsdictschema_version: 1.0.0
software:
dictname: tskitversion: 0.5.8
parameters:
dictcommand: simplifyTODO: add simplify parameters
environment:
dict
os:
dictsystem: Linuxnode: node2 release: 5.15.0-58-generic version: #64-Ubuntu SMP Thu Jan 5 11:43:13 UTC 2023 machine: x86_64
python:
dictimplementation: CPythonversion: 3.11.9
libraries:
dict
kastore:
dictversion: 2.1.1 |
| 05 September, 2024 at 11:49:43 PM | tsinfer | 0.3.3 | infer |
Detailsdictschema_version: 1.0.0
software:
dictname: tsinferversion: 0.3.3
parameters:
dictmismatch_ratio: Nonecommand: infer
environment:
dict
libraries:
dict
zarr:
dictversion: 2.17.2
numcodecs:
dictversion: 0.12.1
lmdb:
dictversion: 1.4.1
tskit:
dictversion: 0.5.8
os:
dictsystem: Linuxnode: node2 release: 5.15.0-58-generic version: #64-Ubuntu SMP Thu Jan 5 11:43:13 UTC 2023 machine: x86_64
python:
dictimplementation: CPython
version:
list311 9 |
Same stuff as before
# the last index is a simply a 2 step starting from position 1
estsfs_diversity = ts_estsfs.diversity(windows=create_windows(ts_estsfs))[1::2]
estsfs_diversity[:10]
array([0.42881531, 0.48983504, 0.49397942, 0.27006834, 0.45935871,
0.24743806, 0.14849395, 0.3905915 , 0.46073859, 0.4897602 ])
Are this values similar to the values calculated using VCFtools?
np.isclose(estsfs_diversity, vcftools_diversity["PI"], atol=1e-6).all()
np.True_
So nucleotide diversity is the same in both cases (using the REF as ancestral allele and using the est-sfs output as ancestral allele). Calculate diversity using branch:
# the last index is a simply a 2 step starting from position 1
estsfs_diversity_branch = ts_estsfs.diversity(mode='branch', windows=create_windows(ts_estsfs))[1::2]
estsfs_diversity_branch[:10]
array([374.15499399, 374.15499399, 374.15499399, 308.89499625,
312.0500738 , 314.27230249, 292.80638906, 294.28489121,
482.83633406, 482.83633406])
Try to plot the tow different diversities with vcftools output:
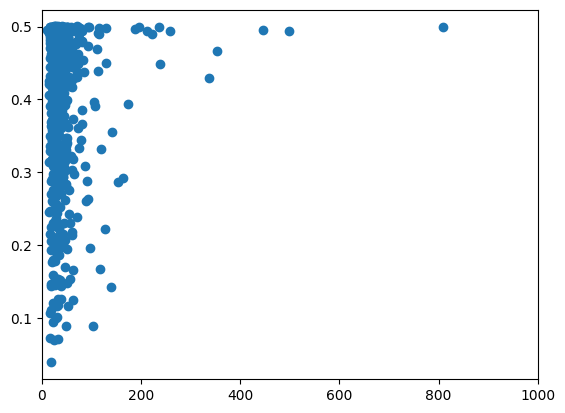
plt.scatter(estsfs_diversity, vcftools_diversity["PI"])
<matplotlib.collections.PathCollection at 0x7f6f3d5cde50>
plt.scatter(estsfs_diversity_branch, vcftools_diversity["PI"])
plt.xlim(0, 1000)
(0.0, 1000.0)
Compara output as ancestral alleles#
Calculate nucleotide diversity using the tree files generated by the pipeline
using the compara approach as ancestral alleles.
ts_compara = tskit.load(str(get_project_dir() / "results-compara/background_samples/tsinfer/SMARTER-OA-OAR3-forward-0.4.10.focal.26.trees"))
ts_compara
|
|
|
|---|---|
| Trees | 496 |
| Sequence Length | 44 077 779 |
| Time Units | generations |
| Sample Nodes | 11 478 |
| Total Size | 89.9 MiB |
| Metadata |
dict |
| Table | Rows | Size | Has Metadata |
|---|---|---|---|
| Edges | 1 838 362 | 56.1 MiB | |
| Individuals | 5 739 | 353.1 KiB | ✅ |
| Migrations | 0 | 8 Bytes | |
| Mutations | 524 747 | 18.5 MiB | |
| Nodes | 19 188 | 932.4 KiB | ✅ |
| Populations | 202 | 4.7 KiB | ✅ |
| Provenances | 4 | 2.4 KiB | |
| Sites | 657 | 33.5 KiB | ✅ |
| Provenance Timestamp | Software Name | Version | Command | Full record |
|---|---|---|---|---|
| 06 September, 2024 at 02:30:20 AM | tsdate | 0.1.7 | inside_outside |
Detailsdictschema_version: 1.0.0
software:
dictname: tsdateversion: 0.1.7
parameters:
dictmutation_rate: 1e-08recombination_rate: None time_units: None progress: None population_size: 10000.0 eps: 1e-06 outside_standardize: True ignore_oldest_root: False probability_space: logarithmic num_threads: None cache_inside: False command: inside_outside
environment:
dict
os:
dictsystem: Linuxnode: node1 release: 5.15.0-58-generic version: #64-Ubuntu SMP Thu Jan 5 11:43:13 UTC 2023 machine: x86_64
python:
dictimplementation: CPythonversion: 3.11.9
libraries:
dict
tskit:
dictversion: 0.5.8 |
| 06 September, 2024 at 02:25:05 AM | tsdate | 0.1.7 | preprocess_ts |
Detailsdictschema_version: 1.0.0
software:
dictname: tsdateversion: 0.1.7
parameters:
dictminimum_gap: 1000000remove_telomeres: True filter_populations: False filter_individuals: False filter_sites: False
delete_intervals:
listlist0209048.0 list44004282.044077779.0 command: preprocess_ts
environment:
dict
os:
dictsystem: Linuxnode: node1 release: 5.15.0-58-generic version: #64-Ubuntu SMP Thu Jan 5 11:43:13 UTC 2023 machine: x86_64
python:
dictimplementation: CPythonversion: 3.11.9
libraries:
dict
tskit:
dictversion: 0.5.8 |
| 06 September, 2024 at 02:25:01 AM | tskit | 0.5.8 | simplify |
Detailsdictschema_version: 1.0.0
software:
dictname: tskitversion: 0.5.8
parameters:
dictcommand: simplifyTODO: add simplify parameters
environment:
dict
os:
dictsystem: Linuxnode: node1 release: 5.15.0-58-generic version: #64-Ubuntu SMP Thu Jan 5 11:43:13 UTC 2023 machine: x86_64
python:
dictimplementation: CPythonversion: 3.11.9
libraries:
dict
kastore:
dictversion: 2.1.1 |
| 06 September, 2024 at 02:25:00 AM | tsinfer | 0.3.3 | infer |
Detailsdictschema_version: 1.0.0
software:
dictname: tsinferversion: 0.3.3
parameters:
dictmismatch_ratio: Nonecommand: infer
environment:
dict
libraries:
dict
zarr:
dictversion: 2.17.2
numcodecs:
dictversion: 0.12.1
lmdb:
dictversion: 1.4.1
tskit:
dictversion: 0.5.8
os:
dictsystem: Linuxnode: node1 release: 5.15.0-58-generic version: #64-Ubuntu SMP Thu Jan 5 11:43:13 UTC 2023 machine: x86_64
python:
dictimplementation: CPython
version:
list311 9 |
Calculate nucleotide diversity as before:
# the last index is a simply a 2 step starting from position 1
compara_diversity = ts_compara.diversity(windows=create_windows(ts_compara))[1::2]
compara_diversity[:10]
array([0.42881531, 0.48983504, 0.49397942, 0.27006834, 0.45935871,
0.24743806, 0.14849395, 0.3905915 , 0.46073859, 0.4897602 ])
Are this values similar to the values calculated using VCFtools?
np.isclose(compara_diversity, vcftools_diversity["PI"], atol=1e-6).all()
np.True_
So nucleotide diversity is the same in both cases (using the REF as ancestral allele and using the compara as ancestral allele). Calculate diversity using branch:
# the last index is a simply a 2 step starting from position 1
compara_diversity_branch = ts_compara.diversity(mode='branch', windows=create_windows(ts_compara))[1::2]
compara_diversity_branch[:10]
array([480.63266349, 363.72632856, 356.52374401, 376.91612567,
381.58518179, 415.57735315, 419.10475683, 419.5249502 ,
419.5249502 , 419.5249502 ])
Try to plot the tow different diversities with vcftools output:
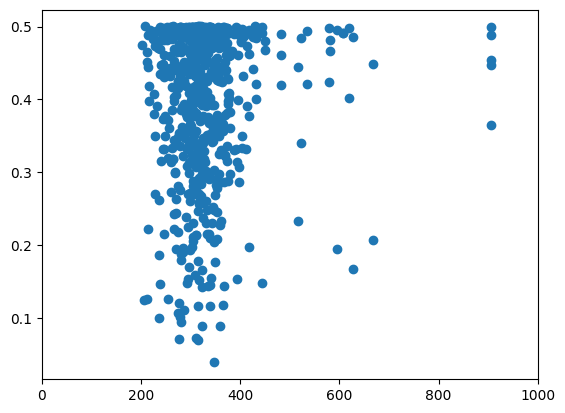
plt.scatter(compara_diversity, vcftools_diversity["PI"])
<matplotlib.collections.PathCollection at 0x7f6f3ec6e5d0>
plt.scatter(compara_diversity_branch, vcftools_diversity["PI"])
plt.xlim(0, 1000)
(0.0, 1000.0)